

Summer Training Exercises
Go Back
Section - 1
Sequence and Genome Analysis
Various Genome Databases:
NCBI Genome Browser
http://www.ncbi.nlm.nih.gov/genome
Ensembl Genome Browser
http://www.ensembl.org/index.html
UCSC Genome Browser
http://hgdownload.cse.ucsc.edu/downloads.html
GENSCAN
GENSCAN is a program to identify complete gene structures in genomic DNA. It is a GHMM-based program that can be used to predict the location of genes and their exon-intron boundaries in genomic sequences from a variety of organisms. It also predicts peptide sequence of predicted genes of given genomic DNA.
Steps:
- Open GenScan webpage: http://genes.mit.edu/GENSCAN.html
- Select 'Organism' - can select DB organism according to organism of input DNA sequence.
- Set 'Suboptimal exon cutoff' value - use 0.5 for better prediction
- Choose 'Print Option' for only peptide or both peptide & cds prediction.
- Browse or paste genomic DNA sequence (less than 1mb data)
- Click on 'Run GenScan' button for the prediction results.
Terms used in Results of GenScan:
Gn.Ex : gene number, exon number (for reference)
Type : Init = Initial exon (ATG to 5' splice site)
Intr = Internal exon (3' splice site to 5' splice site)
Term = Terminal exon (3' splice site to stop codon)
Sngl = Single-exon gene (ATG to stop)
Prom = Promoter (TATA box / initation site)
PlyA = poly-A signal (consensus: AATAAA)
S : DNA strand (+ = input strand; - = opposite strand)
Begin : beginning of exon or signal (numbered on input strand)
End : end point of exon or signal (numbered on input strand)
Len : length of exon or signal (bp)
Fr : reading frame (a forward strand codon ending at x has frame x mod 3)
Ph : net phase of exon (exon length modulo 3)
I/Ac : initiation signal or 3' splice site score (tenth bit units)
Do/T : 5' splice site or termination signal score (tenth bit units)
CodRg : coding region score (tenth bit units)
P : probability of exon (sum over all parses containing exon)
Tscr : exon score (depends on length, I/Ac, Do/T and CodRg scores)
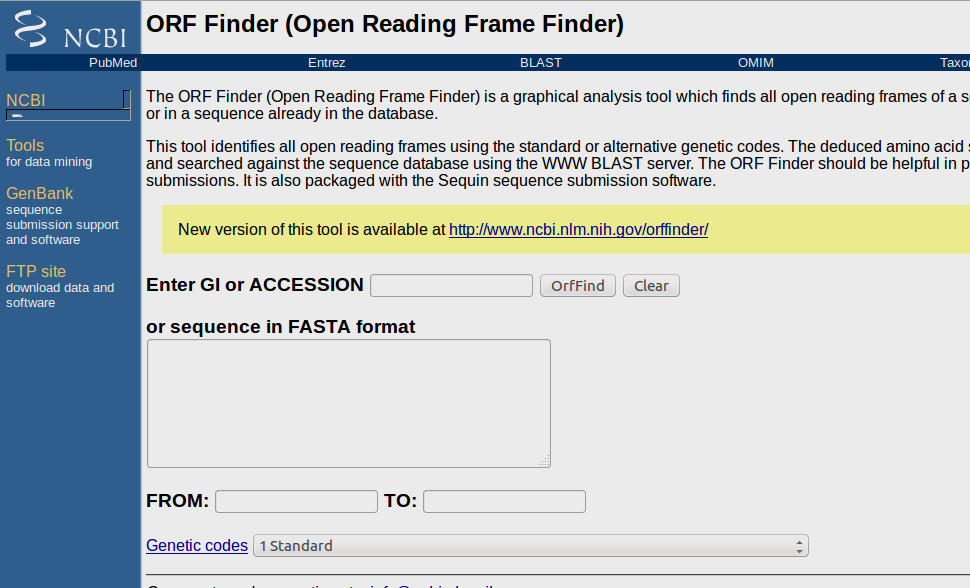
ORF Finder
The ORF Finder (Open Reading Frame Finder) is a graphical analysis tool which finds all open reading frames of a selectable minimum size in a user's sequence or in a sequence already in the database.
This tool identifies all open reading frames using the standard or alternative genetic codes.
The deduced amino acid sequence can be saved in various formats and searched against the sequence database using the WWW BLAST server. The ORF Finder should be helpful in preparing complete and accurate sequence submissions. It is also packaged with the Sequin sequence submission software. GENSCAN is a program to identify complete gene structures in genomic DNA. It is a GHMM-based program that can
be used to predict the location of genes and their exon-intron boundaries in genomic sequences from a variety
of organisms.
It also predicts peptide sequence of predicted genes of given genomic DNA.
Steps:
- Open ORF Finder webpage: http://www.ncbi.nlm.nih.gov/gorf/gorf.html
- Paste sequence in FASTA format or provide accesion no of sequence.
- provide the nucleotide range if needed
- Specify genetic code.
- Click on 'Run Orffind' button for the prediction results.
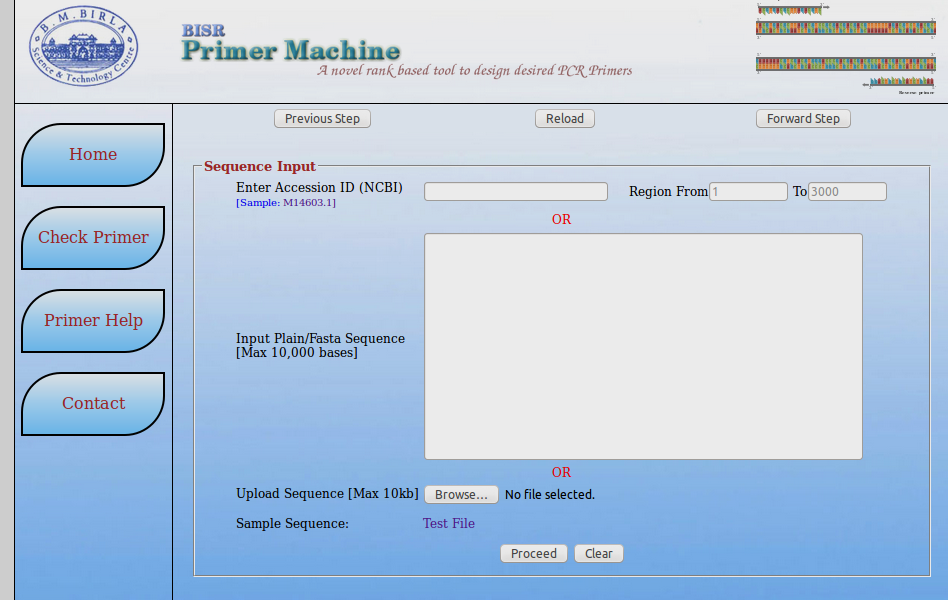
BISR Primer Machine
Steps:
- Open BISR Primer webpage: http://bioinfo.bisr.res.in/cgi-bin/project/primer/index.cgi
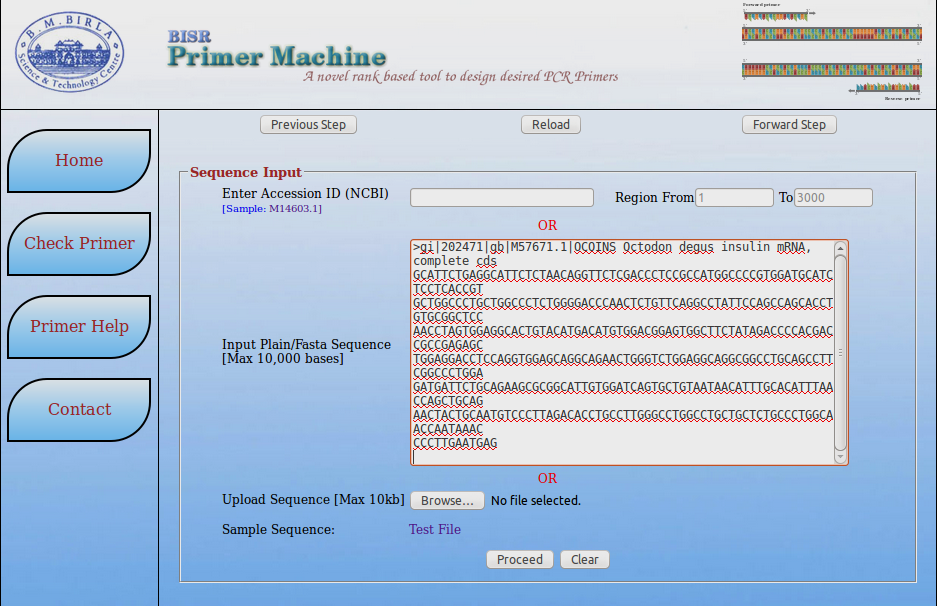
- Paste sequence in FASTA format or provide accesion no of sequence.
- Provide the nucleotide range if required
- You can also take the test file if you are checking it for use.
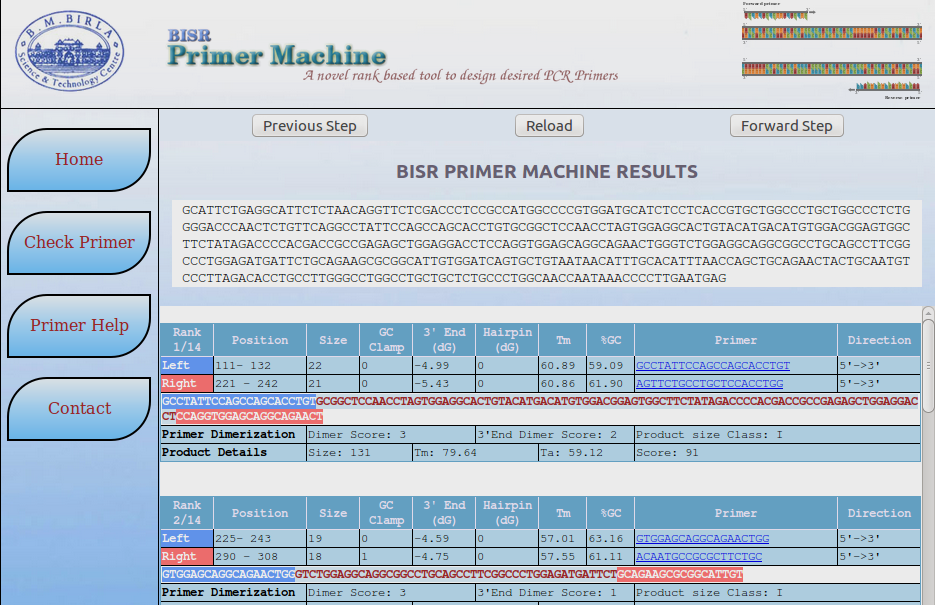
- Click on 'Proceed' button for the prediction results.
- Result will provide the positions of nucleotide in given sequence on the basis of ranks.
VISTA- Tools for Comparative Genomics
-
VISTA is a comprehensive suite of programs and databases for comparative analysis of genomic sequences.
- mVISTA- Align and compare your sequences from multiple species
- gVISTA- Compare your sequences against whole-genome assemblies.
- wgVISTA- Align pair of sequences up to 10Mb long (finished or draft) including microbial whole-genome assemblies.
- Open VISTA tool from the link http://genome.lbl.gov/vista/index.shtml
- At the first page you will be asked to identify the number of genomic sequences you want to analyze. Entering this number and clicking "submit" will take you to the main submission page which will contain the number of fields corresponding to the number of sequences you entered (can process up to 100 sequences).
- It will ask for email address so that it can notify you when the results are ready.
- Upload query sequence in Fasta format using "Browse" button (previously saved sequence).
- You can specify its GenBank accession number, which will be used to automatically retrieve the sequence from the GenBank database and process on our server.
- Click Submit.
There are two ways of using VISTA: either submit your own and alignments for analysis or examine pre-computed whole-genome alignments of different species.
-
There are three tools in VISTA for genome analysis-