

Go Back
Section - 1
Sequnce Similartity and Alignment: BLAST, FASTA and Clustal Omega
Lab 1: Local Alignment -BLAST
Basic Local Alignment Search Tool (BLAST) is a dynamic programming algorithm for sequence alignment. It is based on smith-waterman algorithm, means on the concept of local alignment. For an alignment blast uses a query sequence which is taken for the alignment and one or more subject seqeunces on which the query sequence is aligned.
| Nucleotide Blast | Search a nucleotide database using a nucleotide query |
| protein blast | Search protein database using a protein query |
| blastx | Search protein database using a translated nucleotide query |
| tblastn | Search translated nucleotide database using a protein query |
| tblastx | Search translated nucleotide database using a translated nucleotide query |
STEPS:
- Open BLAST tool
Link is :http://blast.ncbi.nlm.nih.gov/Blast.cgi - Click on nucleotide blast (blastn).
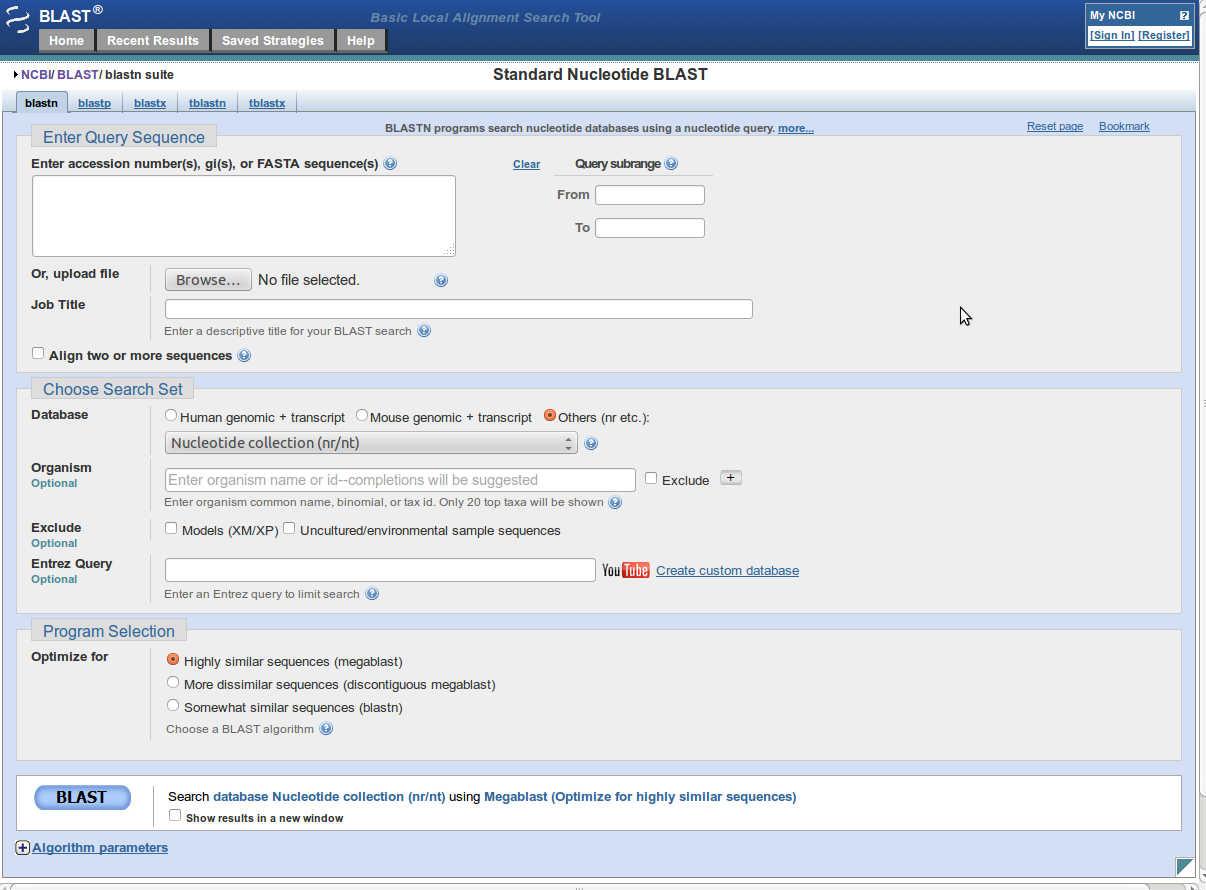
- Enter fasta sequence or GI number or accession number of query sequence into 'Enter Query Sequence'.
- You can set further parameters for blast.
- The parameters set there are default, if any change is made in these parameters, a light yellow strip is displayed under that parameter.
- After setting up all the parameters click on 'BLAST' button to run blast.
- This will take to the result page in some time.
Result page of BLAST
Result inculudes graphics summary, descriptions, dot plots, alignment, scores etc.
In graphics summary result is shown as different color showing a range of percent similarity score.Towards
black color, less alignment score means less similarity and towards red color more alignment score means
more similarity.
The very first red strip below the range strip with numbers below show the query sequence with its length.
After that all colored lines show alignment of query sequence with various (subject) sequences available in
database.
After that description of subject sequences are given. It includes name of subject sequence, scores,
indentity with query, e-value and the link of subject sequence.
After 'Description' alignment of query sequence with various subject sequences are given. It shows base to
base alignment with matches, mismatches and gaps as well.
Lab 2: Global Alignment -FASTA
FASTA (pronounced as fast- aye) is an another tool for sequence similarity search analysis. It makes alignment
of query protein sequence against protein database's sequences globally. It is based on needleman and wunsch
algorithm.
Two another tools of FASTA suit are FASTX and FASTY. These tools are used for the nucleotide query
sequences.
| Program Name | Description | Abbreviation |
|---|---|---|
| FASTA | Scan a protein or DNA sequence library for similar sequences. | fasta |
| FASTX | Compare a DNA sequence to a protein sequence database, comparing the translated DNA sequence in forward and reverse frames. | fastx |
| FASTY | Compare a DNA sequence to a protein sequence database, comparing the translated DNA sequence in forward and reverse frames. | fasty |
| SSEARCH | Compare a protein or DNA sequence to a sequence database using the Smith-Waterman algorithm. | ssearch |
| GGSEARCH | Compare a protein or DNA sequence to a sequence database using a global alignment (Needleman-Wunsch) | ggsearch |
| GLSEARCH | Compare a protein or DNA sequence to a sequence database with alignments that are global in the query and local in the database sequence (global-local). | glsearch |
STEPS:
- Open page of FASTA tool
Link is :http://www.ebi.ac.uk/Tools/sss/fasta/ - Select protein database, against the alignment will be performed..
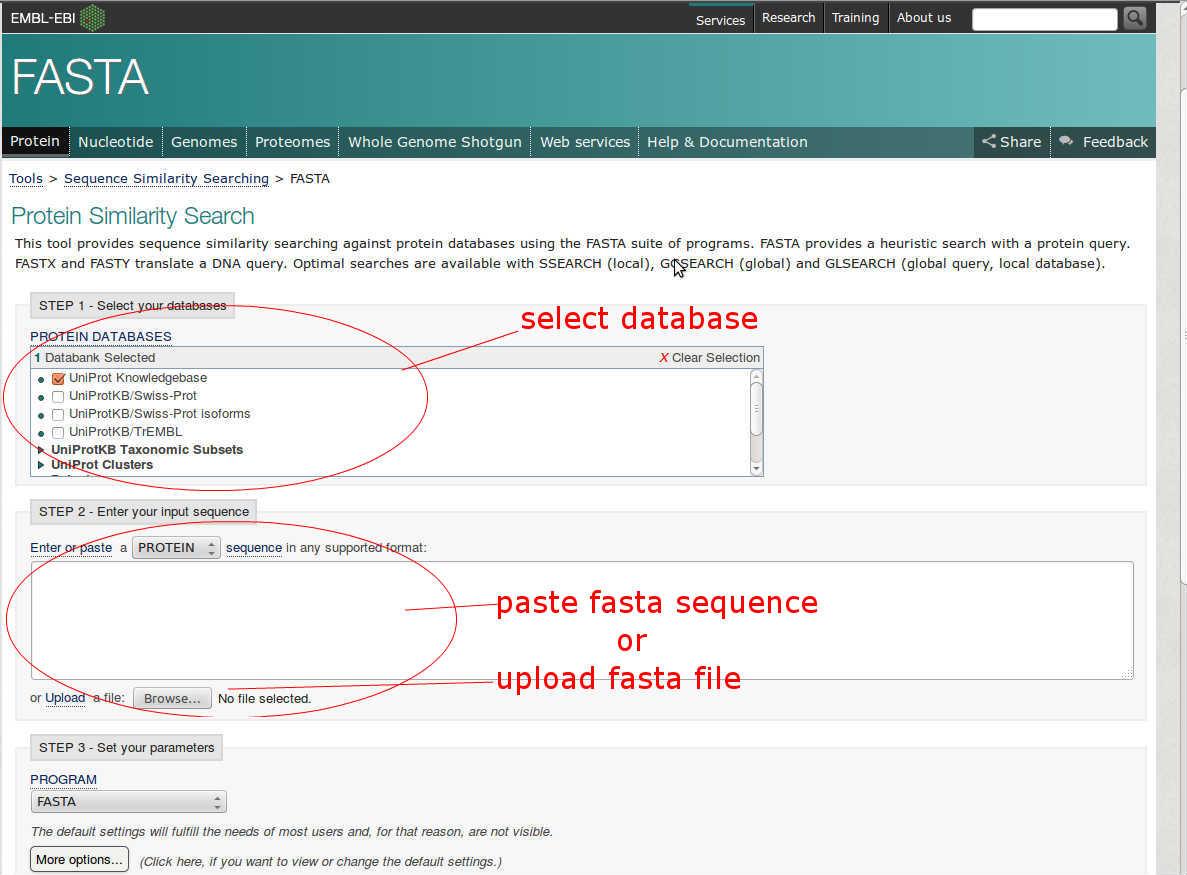
- Then input the fasta sequence of protein.
- There are various parameters given also to make changes in alignment result. You can set them to default.
- Now submit the job and get the alignment result.
Result page of FASTA
The result page of FASTA shows various information regarding to alignment.
Summary Table:
In this portion you will get many subject sequences (searched from the selected database) that have been
highly aligned to the query sequence. It also has information of subject sequence lenght, alignment
score, percent indentities, percent positives (similarity), and e-value.
You can show alignment of query and subject sequecne by clicking on 'Show' button below the alignment
in left side division box.
Tools Output:
It shows the same results as summary table but not in graphically form.
Visual Output:
In this part of result you will get alignment of query and subject sequences in form of colored line.
Changes in color are defined as percent similarity according to e-value.
Functional Prediction:
In this page you will get the alignment of functional part of query sequence with function part of other
proteins. Mainly these fucntional part in protiens are known as secondary structures. Means this is the
alignment result of secondary structure elements.
Submission Details:
This page contains all the parameters details submitted for the alignent.
BLAST and FASTA tools are used for pairwise sequence alignment.
Lab3: Clustal Omega
Clustal-Omega is a general purpose multiple sequence alignment (MSA) program for protein and DNA/RNA. It produces high quality MSAs and is capable of handling data-sets of hundreds of thousands of sequences in reasonable time. It uses seeded guide trees and HMM profile-profile techniques to generate alignments. Minimum three sequences are taken for the MSA.
STEPS:
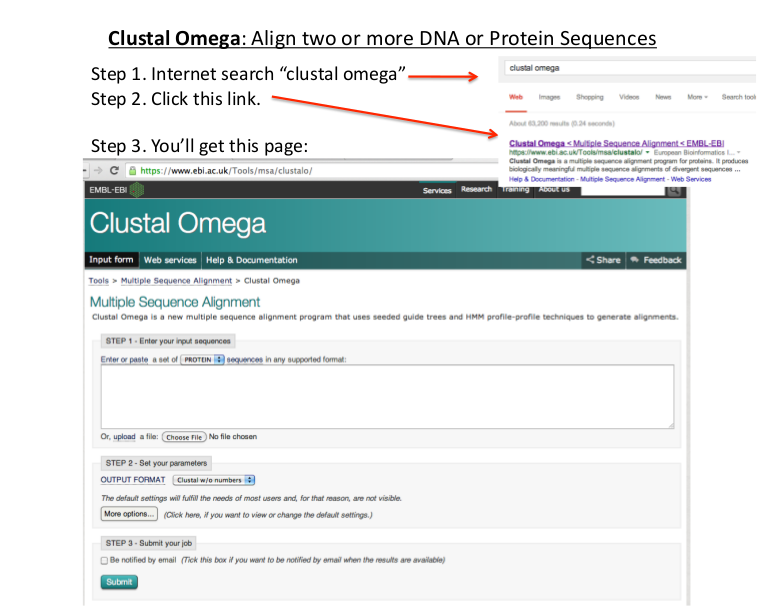
- To perform MSA on Clustal Omega open the following link
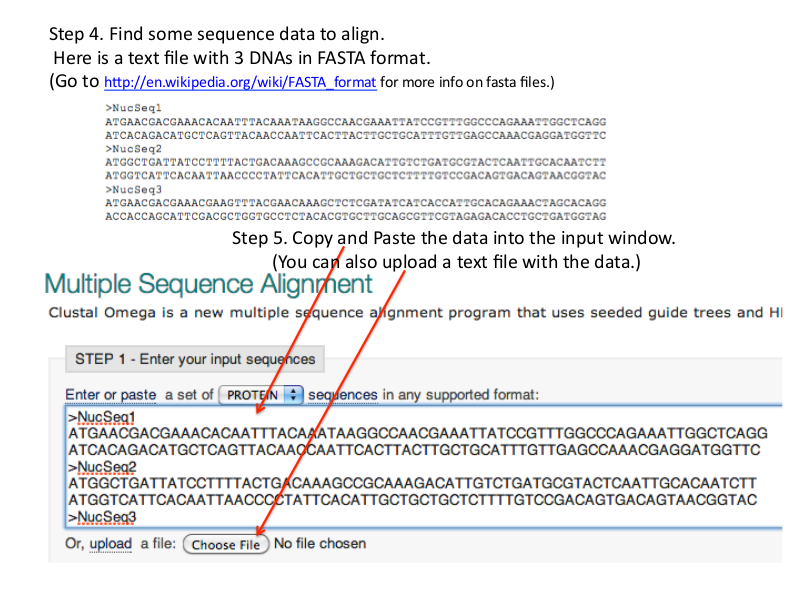
http://www.ebi.ac.uk/Tools/msa/clustalo/ - Enter sequences into Input Sequences Box.
- Default parameters can be set to change the result of alignment.
- Click to submit button to view the alignment result. It will take some time to produce the result.
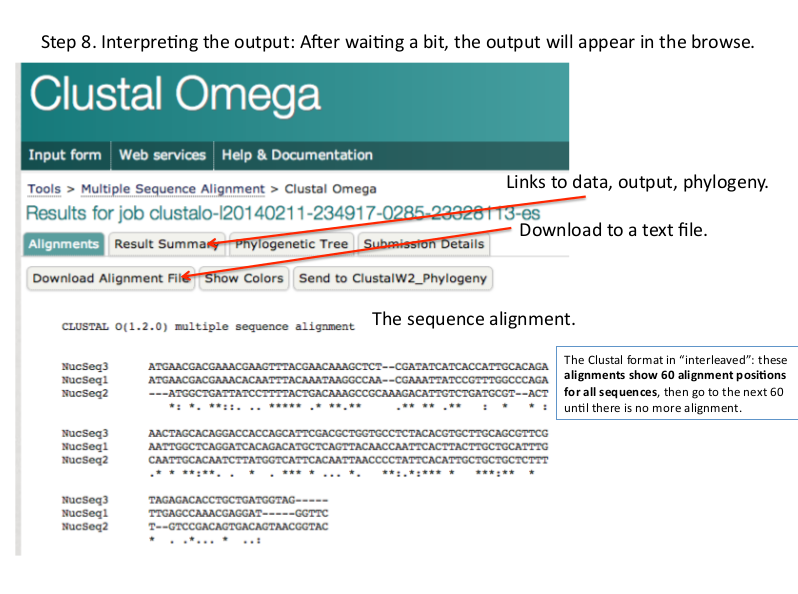
Results of Multiple Sequcnce Alignment (Clustal Omega)
Alignment:
It shows the base per base alignment of each all sequences. Matches, mismatches, gaps are shown their.
There are three types of symbols given below the alignment of some bases:
* [asterisk] It show strong alignment or identical bases in all sequences.
: [colon] It shows strong alignment of similar class bases.
. [period] It shows weak alignment of similar class bases.
Result summary:
It provides alignment result in various format. You can download your msa result as text file from here. You can also get phylogenetic tree file from this page.
Phylogenetic tree
It shows phylogeny among sequences and displays tree.
Submission Details:
This page contains all the parameters details submitted for the alignent.