

Go Back
Prelude:
- Download the repositories from various sources, viz. GEO, Repositive and other genebanks
- FTPing the references
- Compare the paired-end, mate-pair and single-end reads
- making batch scripts for next generation sequencing
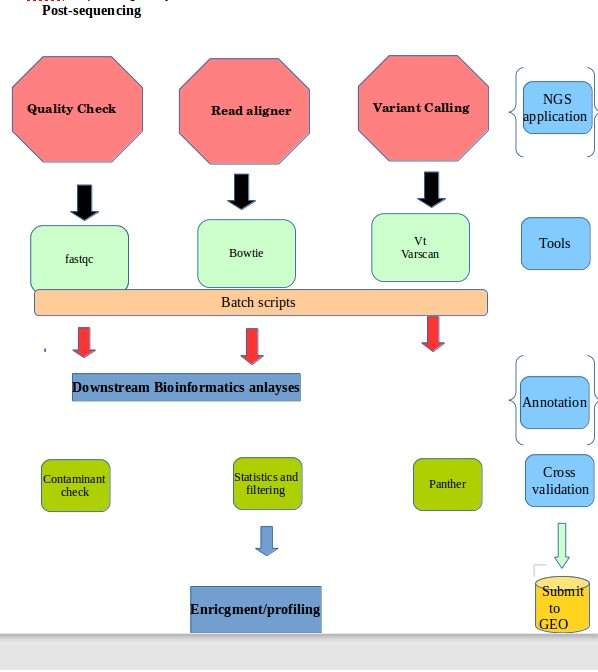
- Overview of all NGS tools, viz. Checking quality mappable reads using Fastqc, Variant calling using vt and varscan, GATK and online tools
- Overview of Galaxy, Git and bitbucket/Atlassian
************************************************************************
Example with Exome sequening data
All users by default are logged into their respective user accounts, viz. Test1, test2, test3 and test4
To login, open the terminal and first ssh 192.168.1.18, please enter username test1/test2/test3/test4 (password shall be informed to you later)
You will be at
/home/test1
Please use your working directory as /home/bioinfo
So to move to that folder, simply type cd ../bioinfo
All your programs/commands/scripts are available in that folder
Please refer sample batch script for exome sequencing here.
*******************************************************************
#Indexing already done using bowtie2, BWA and samtools: ../data/hg38/
#All scripts and commands are to be run from /home/prash/share/analyses/Expipe
#fastqc already one for all samples. Pl check the folder out/
#../bowtie2-master/./bowtie2 -x ../data/hg38/hg38 -1
../../../../admin1/share/km/raw_data/sample_R1.fq.gz -2
../../../../admin1/share/km/raw_data/sample_R2.fq.gz > sample.sam
#samtools import ../data/hg38/hg38.fa sample.sam sample.bam
#samtools sort sample.bam -o sample.sorted.bam
#samtools index sample.sorted.bam sample.sorted.bam.bai
#samtools merge sample.merged.bam sample.sorted.*
#samtools mpileup sample.sorted.bam -o sample.mpileup.bam
#java -jar varscan/varscan.jar mpileup2snp sample.mpileup.bam > sample.mpileup.snps
#java -jar varscan/varscan.jar mpileup2indel sample.mpileup.bam > sample.mpileup.indels
#java -jar ../varscan/varscan.jar filter sample.mpileup.snps >sample.mpileup.snps.filter
#java -jar ../varscan/varscan.jar readcounts sample.mpileup.bam >sample.mpileup.readcounts
#samtools mpileup -uf ../data/hg38/hg38.fa sample.sorted.bam
| bcftools view - > sample.var.raw.bcf
##bcftools view sample.var.raw.bcf | vcfutils.pl varFilter -D100 > sample.var.flt.vcf
#samtools calmd -Abr sample.sorted.bam ../data/hg38/hg38.fa > sample.baq.bam
samtools mpileup -uf ../data/hg38/hg38.fa sample.sorted.bam | bcftools call -c -v -o sample.vcf.gz
#samtools mpileup -go sample.bcf -f ../data/hg38/hg38.fa sample.bam
#bcftools call -vmO z -o sample.vcf.gz sample.bcf
#Preparing VCF for querying and indexing using tabix:
#tabix -p vcf sample.vcf.gz
#preparing graphs and stats:
#bcftools stats -F ../data/hg38/hg38.fa -s sample.vcf.gz > sample.vcf.gz.stats\
#Plots in directory plots
#plot-vcfstats -p plots/ sample.vcf.gz.stats
#Data Filtering:
#bcftools filter -O z -o sample_filtered.vcf.gz -s LOWQUAL -i'%QUAL>10' sample.vcf.gz