

Go Back
Section - II: Protein Structure Prediction: Homology Modeling
Lab 1: Homology Modeling using Swissmodel
Steps:
- Take a protein sequence in FASTA format whose structure is to be modelled. This is our 'Target Sequence'.
- Now go to the webpage http://swissmodel.expasy.org/

- Paste target sequence into swiss model workspace in FASTA format
(you can also upload target sequence )
you can provide a Project title and email-id-
You have 2 options now you can either search for templates or build models directly
- you can see result after a while
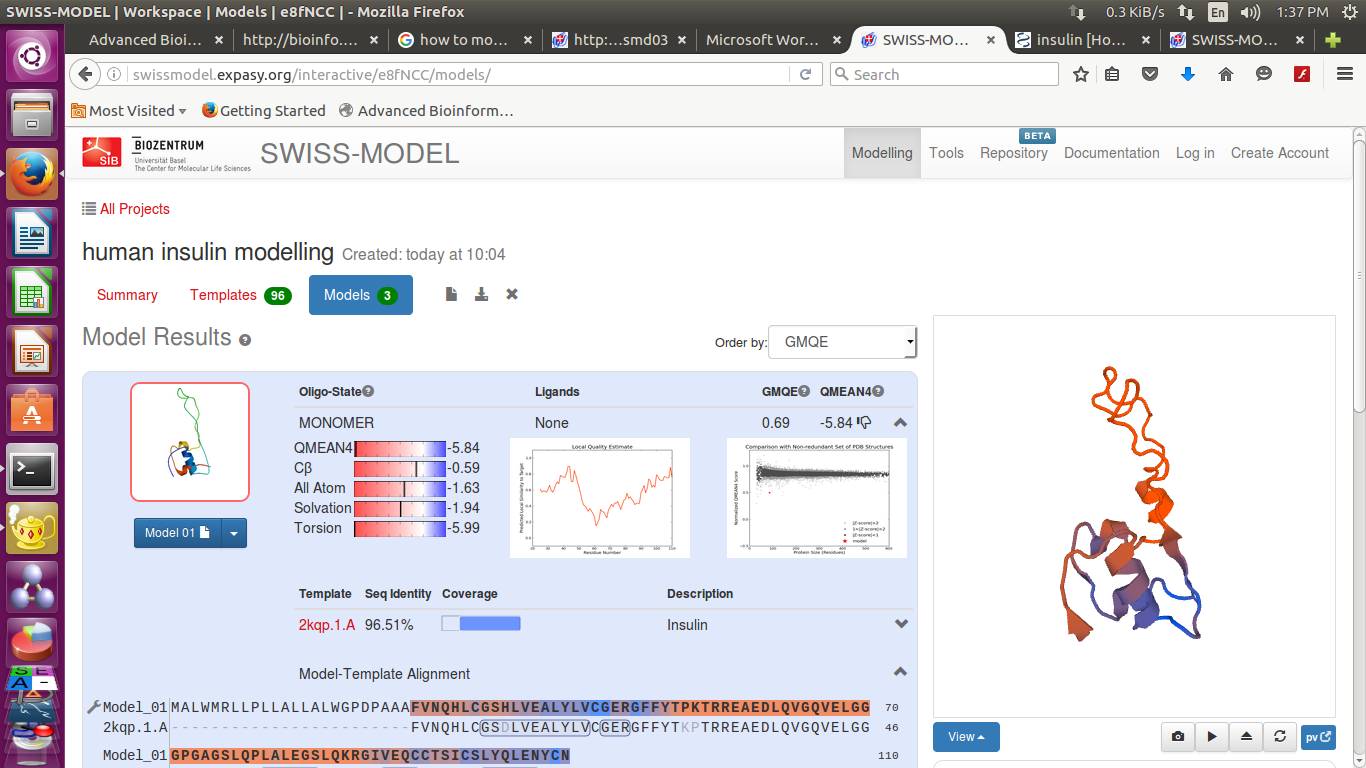
- We can also download the detailed results
The Structure Analysis and Verification Server version 4
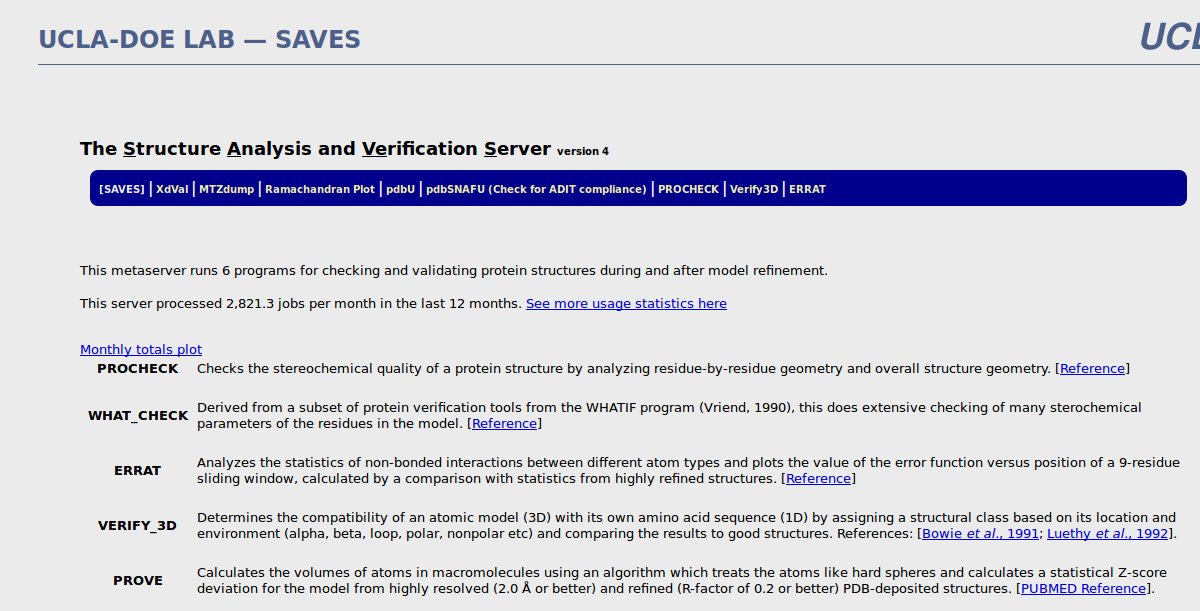 This metaserver runs 6 programs for checking and validating protein structures during and after model refinement.
This metaserver runs 6 programs for checking and validating protein structures during and after model refinement.
PROCHECK : Checks the stereochemical quality of a protein structure by analyzing residue-by-residue geometry and overall structure geometry. [Reference]
WHAT_CHECK : Derived from a subset of protein verification tools from the WHATIF program (Vriend, 1990), this does extensive checking of many sterochemical parameters of the residues in the model. [Reference]
ERRAT : Analyzes the statistics of non-bonded interactions between different atom types and plots the value of the error function versus position of a 9-residue sliding window, calculated by a comparison with statistics from highly refined structures. [Reference]
VERIFY_3D : Determines the compatibility of an atomic model (3D) with its own amino acid sequence (1D) by assigning a structural class based on its location and environment (alpha, beta, loop, polar, nonpolar etc) and comparing the results to good structures. References: [Bowie et al., 1991; Luethy et al., 1992].
PROVE : Calculates the volumes of atoms in macromolecules using an algorithm which treats the atoms like hard spheres and calculates a statistical Z-score deviation for the model from highly resolved (2.0 Å or better) and refined (R-factor of 0.2 or better) PDB-deposited structures. [PUBMED Reference].
CRYST1 record matches : We take the CRYST1 record and search the entire PDB for matches and report these as possibly similar structures.
Ramachandran Plot : We produce an interactive Ramachandran plot. Also a standalone server linked above.
WedMol Viewer : We provide a structure viewer in the web page, although not all browsers support it. We are working to pvoride a more robust viewer - Open SAVES SERVER webpage: http://services.mbi.ucla.edu/SAVES/
- Upload the PDB file.
- RUN all programs
Press the button 'build model'.
Swissmodel provide 3 best results according to their score
It provide- Sequence identity, Alignment and Structure